Defective bicarbonate reabsorption in Kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion.
The kidneys excrete the daily acid load mainly by generating and excreting ammonia but the underlying molecular mechanisms are not fully understood. This study evaluated the role of the inwardly rectifying potassium channel subunit Kir4.2 (Kcnj15) in this process.
Friday 31 January 2020 - 12h15 to 13h15 - Bugnon 27 - Seminar room Lipari
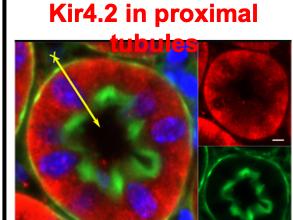
In mice, Kir4.2 was present exclusively at the basolateral membrane of proximal tubular cells and the invalidation of Kcnj15 caused a hyperchloremic metabolic acidosis associated with a reduced threshold for HCO3- in the absence of a generalized proximal dysfunction. Urinary NH4+ excretion rates in Kcnj15-deleted mice were inappropriate to acidosis under basal and acid-loading conditions, and not related to a failure to acidify urine or a reduced expression of ammonia transporters in the collecting duct. In contrast, the expression of key proteins involved in ammonia metabolism and secretion by proximal cells was inappropriate in Kcnj15-deleted mice. In addition, Kcnj15 deletion depolarized the proximal cell membrane by decreasing the barium-sensitive component of the potassium conductance and caused an intracellular alkalinization.
We conclude that the Kir4.2 potassium channel subunit is a new regulator of proximal ammonia metabolism and the renal consequences of its loss of function in mice support the proposal for KCNJ15 as a molecular basis for human isolated proximal renal tubular acidosis.
Dr Dmitri Firsov